A blog about being too bendy. To raise awareness of Ehlers Danlos Syndrome.
Being diagnosed with EDS.
(1999-2005).
After my exercise programme I was much more active and feeling better, but I still struggled with some very basic tasks. When you walk, the movement of the muscles in your legs helps pump blood around your body, but when you’re stationary this doesn’t happen. So although I could walk over 70 miles in a week, if I sat upright in a chair and chatted to a friend within a few minutes I would feel light headed and would feel blood collecting in my feet. My concentration would also be poor, a condition known as ‘brain fog’ (ref), caused by lack of blood to the brain. I discussed these problems with Professor Mathias. He suggested that to make further progress I should try exercises that challenged my cardiovascular system more than walking.
I started gradually trying more strenuous exercises, but quickly got a thigh strain and hurt my back. I made no progress for months due to frequent muscle strains. It wasn’t until I found a physiotherapist with experience of rehabilitation that I started to improve again. Nine months later I’d gained weight, I was walking every day, and doing a range of other exercises.
It seemed to be time to challenge my body more, so I started using an exercise bike. I began slowly but quickly got another thigh strain. It was incredibly frustrating. This pattern carried on for a couple of years. I worked very hard to get stronger but instead I picked up worse and worse injuries. Slowly it became normal for me to be limping. Sometimes I could barely walk at all.
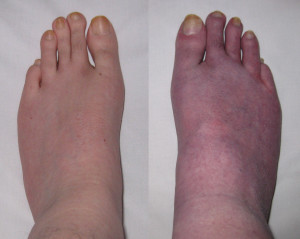
My feet, before and after 10 minutes standing, showing venous pooling.
As I gradually became less capable of exercise, as a consequence my POTS symptoms also got worse. In 2001 I went into hospital for some cardiovascular tests. During a ward round Professor Mathias asked me to stand up so that a group of students could watch venous pooling occur (venous pooling is when blood collects in the lower legs turning the person’s feet purple). So I stood in my underwear while his students stared at my feet. Feeling embarrassed, I tried to lighten the moment by apologising for my ugly feet and elongated toes. A couple of hours later some doctors returned and asked to take some measurements.
I won’t forget the next ward round! Professor Mathias said ‘We think you have a connective tissue disorder, either Marfan Syndrome or Ehlers Danlos Syndrome. We think it could be causing your POTS symptoms’. He told me I was the first patient his department had found with a connective tissue disorder and POTS, and they planned to see if any of their other patients have both conditions.
The measurements showed I have a ‘Marfanoid Habitus’, a body shape common in people with a connective tissue disorder. Features include a tall slender physique, with long arms, legs, fingers and toes. I have all those characteristics, plus my arm-span is greater than my height which is a key indicator. At 6 foot 1 inch, I’m oddly tall for my family, 5 inches taller than my parents, and 12 inches taller than my sister.
I was referred to Professor Rodney Grahame at University College Hospital, a rheumatologist and specialist in hypermobility. Thankfully tests on my heart and eyes ruled out Marfan Syndrome, which can cause serious cardiac problems. Professor Grahame found most of my joints were hypermobile (double jointed), some ‘markedly so’. I’d always known my fingers were double jointed, but I wasn’t aware my other joints were. A skin biopsy also found that my skin was hyperelastic (very stretchy). Due to these features, my marfanoid body shape, and a history of joint pain, Professor Grahame diagnosed me with Ehlers-Danlos Syndrome Hypermobility type (now known as hypermobile Ehlers-Danlos Syndrome).
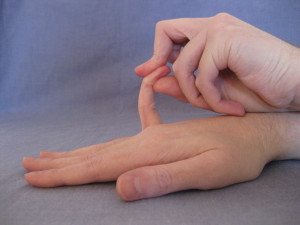
Beighton Finger Test. – One point for each little finger that bends backwards beyond 90 degrees.
Prof. Mathias and Prof. Grahame have since established that 80-90% of POTS patients also have hypermobile Ehlers-Danlos Syndrome. In a healthy person blood vessels constrict slightly on standing to help cope with gravity. In someone with EDS the weak connective tissue in blood vessels causes them to expand under pressure rather than constrict, so blood sinks into the hands and lower legs (venous pooling). The rise in heart rate which patients experience is the body’s attempt to prevent this. (ref).
Being diagnosed with EDS not only explained all the muscle and joint problems I’d been having over the previous few years, but also the numerous injuries I had as a teenager. (My file of old X-rays at the local hospital was so thick I was once asked if I was being bullied or abused!) – Unfortunately it also changed my prognosis for the future. I adopted a pain management approach to my joint problems, which meant taking more pills but fewer physio sessions. I was given a wheelchair for the periods when I couldn’t walk.
It was a big change in approach but over the next few years I managed to have more of a life. I had weekends away and moved into a flat on my own. I also became an uncle which was the biggest highlight, so I had a new buddy to play with.
As I had experienced with POTS, achieving my diagnosis of Ehlers Danlos Syndrome was also detrimentally slow. The most disabling injuries I’ve had occurred before I was diagnosed with EDS. They were caused by trying to get stronger too quickly and have never fully healed. If I’d known I had EDS earlier and been advised how to manage the condition I don’t think I’d be in such a poor state today. But the link between conditions like POTS and Ehlers Danlos Syndrome wasn’t discovered until 1999 (ref). It was noticed in me in 2001 when I’d already been ill for 7 years. It’s frustrating to look back on this, but thankfully progress has been made. Newly diagnosed POTS patients are now tested for EDS straight away, saving them the years of bewildering injuries I experienced.
Ehlers Danlos Syndrome can also effect the digestive system…